Researchers at Zhejiang and Xiamen universities discovered how toxic tau protein clumps reactivate silent DNA to trigger neuron death, and found that blocking a single protein could slow cognitive decline.
Scientists from Zhejiang University, Xiamen University, and collaborating institutions have just published a study in Nature Neuroscience that traces a precise molecular chain of events from tau protein buildup all the way to neuron death. More importantly, they think they’ve spotted a weak link in that chain that could one day be targeted with drugs.
We’ve known for decades that a protein called tau is involved in the disease, but how it kills brain cells has remained a mystery.
From Helpful Protein to Deadly Tangles
Under normal circumstances, tau is actually one of the brain’s good guys. It stabilizes microtubules, which are the internal scaffolding that keeps neurons structurally sound and allows nutrients to travel through them. Without tau doing its job, neurons would collapse from the inside.
In Alzheimer’s disease and a group of related conditions called tauopathies, these Tau proteins misfold and begin clumping together into toxic, thread-like tangles that accumulate inside neurons. These tangles are not just inert debris, they’re actively harmful, and their accumulation drives the progressive death of brain cells that underlies the memory loss and cognitive decline that define the disease.
Treatments that can meaningfully interrupt tau’s toxicity have so far eluded researchers. There have been experimental therapies that tried to reduce tau levels or prevent aggregation, but none have translated cleanly into clinical success.
Part of the problem is that we haven’t had a complete picture of exactly how tau goes from “clumped” to “deadly”. This new study fills in a crucial piece of that picture.
The Molecular Chain Reaction Behind Tau Toxicity
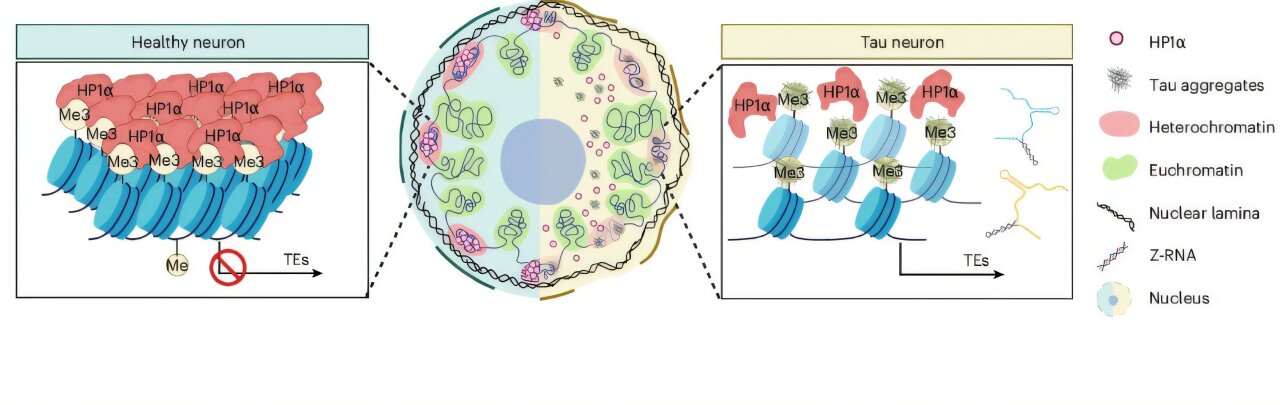
Much of our genome isn’t made up of genes that code for proteins. A significant portion consists of ancient viral remnants and so-called transposable elements. In simple words, they are the sequences that, over billions of years of evolution, have been stitched into our DNA and then, critically, locked away. The cell wraps this DNA tightly into a structure called heterochromatin and the sequences inside are kept inactive.
The Chinese research team asked a specific question:
What happens to this inactive set when tau clumps up inside neurons?
To find out, they worked with a mouse model called PS19, mice genetically engineered to develop abnormal tau aggregation that closely mirrors what happens in human tauopathies. These mice progressively show memory problems and cognitive decline, making them a well-established model for studying the disease.
Researchers found that Tau aggregates have a strong chemical affinity for a specific molecular marker on heterochromatin, a modification called H3K9me3. By latching onto this marker, the tau clumps drew away a critical “guardian” protein called HP1 (heterochromatin protein 1) that normally keeps the vault sealed. Without HP1 on the job, the heterochromatin loosened. The vault cracked open.
“Tau aggregates show a strong affinity for H3K9me3-modified chromatin, effectively sequestering these epigenetic marks from HP1, thereby disrupting the condensation of constitutive heterochromatin”.
— Liu, Wu et al., Nature Neuroscience, 2026
With the vault open, those transposable elements, genetic sequences that the cell had worked hard to silence, were suddenly free to become active. They produced a type of RNA molecule called Z-RNA. And that’s when things went from bad to catastrophic for the neuron.
Z-RNAs are not inherently problematic molecules. But they are a molecular signal that something unusual is happening, normally, the immune system uses them to detect viral infections. Inside neurons undergoing tau-driven heterochromatin disruption, these Z-RNAs triggered the activation of a protein called ZBP1 (Z-DNA-binding protein 1).
ZBP1 is a sensor of cellular stress and danger, and when it gets activated, it can set off a form of programmed cell death called necroptosis.
Unlike apoptosis, where the cell’s orderly, self-contained method of dying, necroptosis is inflammatory. Cells undergoing necroptosis burst, releasing their contents and triggering inflammation in surrounding tissue. For neurons, this is catastrophic and if it’s happening repeatedly across a brain region, the cumulative damage is exactly what drives the progressive cognitive decline seen in Alzheimer’s and related diseases.
The Five-Step Chain Reaction
The researchers were able to trace this process step by step, following the path from tau tangles to the final destruction of neurons. The sequence looks like this:
| Step | What Happens | Why It Matters |
| 1 Tau aggregates form | Misfolded tau proteins clump into toxic tangles inside neurons | The initial event that triggers the entire downstream cascade |
| 2 Heterochromatin disrupted | Tau clumps bind H3K9me3 marks, pulling HP1 away from tightly packed DNA | Breaks the “seal” that keeps dormant DNA silent |
| 3 Transposable elements reactivate | Ancient, normally-silenced genetic sequences begin expressing themselves | Produces Z-RNA molecules that act as a false alarm signal |
| 4 ZBP1 gets activated | Z-RNAs trigger ZBP1, a stress sensor that normally responds to viral RNA | Initiates necroptosis (inflammatory cell death) |
| 5 Neuron death via necroptosis | Neurons undergo inflammatory cell death and release damaging contents | Directly drives neurodegeneration and cognitive decline |
Blocking ZBP1 Improved Memory in Aging Mice
The team didn’t just map the mechanism, they tested whether disrupting it could make a difference. In mice that had only one functional copy of the Zbp1 gene (what scientists call haploinsufficiency, partial knockdown of the protein), aged tau-transgenic mice showed significantly improved cognitive performance compared to controls.
Twenty-four-month-old mice are quite old in rodent terms, roughly comparable to elderly humans, and the fact that reducing ZBP1 activity at this stage still translates to measurable cognitive benefit is encouraging.
It suggests that even late intervention in the cascade might have therapeutic value, rather than only approaches that prevent tau from aggregating in the first place.
There’s also a human data angle that lends this finding credibility beyond the mouse model. When the researchers analyzed gene expression data from human Alzheimer’s patients, they found a consistent pattern, higher ZBP1 expression in excitatory neurons correlated with worse cognitive performance.
This correlation between what the mice showed and what the human data reflected is exactly the kind of cross-validation that makes a potential drug target worth pursuing.
Takeaway: Promising, But Still Early Days
Of course, there’s still a long road between a promising discovery in mice and a treatment that helps people. Neuroscience is full of findings that looked exciting in animal studies but didn’t hold up in human clinical trials. Even the researchers behind this work are careful about their claims. They’re not saying they’ve found a cure. What they’ve done is identify a new mechanism and a potential therapeutic target.
After decades of false starts and promising ideas that didn’t quite pan out, discoveries like this stand out. Not because they immediately change treatment, but because they reveal something new about how the disease works. And there’s something striking about the possibility that ancient pieces of dormant DNA might be helping drive the loss of brain cells in Alzheimer’s disease. It’s the kind of connection few researchers would have predicted, yet it may turn out to be an important part of the story.
Source: Medicalxpress
Publication details
Wei Liu et al, Tau aggregates cause reactivation of transposable DNA elements, leading to Z-RNA–ZBP1-mediated neuronal death, Nature Neuroscience (2026). DOI: 10.1038/s41593-026-02299-9
Frequently Asked Questions
Q1: Why is this research important for Alzheimer’s treatment?
It identifies a specific molecular pathway, tau aggregates disrupting heterochromatin, which activates ZBP1-driven neuron death, that was previously unknown. More critically, it pinpoints ZBP1 as a possible drug target. Most existing Alzheimer’s treatments focus on amyloid plaques, this opens a parallel avenue targeting the mechanism of neuron death itself, which could complement or even surpass current approaches.
Q2: What is ZBP1, and why is it a target?
ZBP1 (Z-DNA-binding protein 1) is a cellular sensor involved in inflammation and programmed cell death. The study found that tau-induced activation of transposable elements can trigger ZBP1, leading to neuron death.
Q3: What are transposable elements, and why do they matter here?
Transposable elements are ancient genetic sequences, sometimes called “jumping genes” or genomic fossils, that make up a large portion of human DNA. Over evolutionary time, the cell learned to silence them by locking them into tightly packed heterochromatin. In this study, tau aggregates interfere with that silencing mechanism, allowing these sequences to become active and produce Z-RNA molecules that ultimately trigger cell death. Their reactivation is a false alarm that turns lethal for neurons.
Q4: Does this research apply only to Alzheimer’s, or to other diseases too?
The mechanism is relevant to the broader family of tauopathies, which includes frontotemporal dementia, progressive supranuclear palsy, and CTE (the brain disease associated with repeated head trauma). All of these involve abnormal tau aggregation in neurons. If ZBP1 inhibition proves effective, it could potentially benefit patients with these conditions as well, significantly expanding the scope of the research’s impact.
Q5: When could this discovery lead to an actual treatment?
The findings are still at the preclinical stage. If future studies confirm the results in humans, therapies targeting the ZBP1 pathway could emerge within the next decade, though timelines for drug development remain uncertain.
Q6: Does this replace existing Alzheimer’s theories?
No. Instead, it adds another layer of understanding. Tau aggregation remains a central feature of Alzheimer’s disease, but this study helps explain one of the ways those aggregates may cause damage inside neurons.